PROVIDENCE, R.I. [Brown University] — Using a computer algorithm that can sift through mounds of genetic data, researchers from Brown University have identified several networks of genes that, when hit by a mutation, could play a role in the development of multiple types of cancer.
The algorithm, called Hotnet2, was used to analyze genetic data from 12 different types of cancer assembled as part of the pan-cancer project of The Cancer Genome Atlas (TCGA). The research looked at somatic mutations — those that occur in cells during one’s lifetime — and not genetic variants inherited from parents. The study identified 16 subnetworks of genes — several of which have not previously received much attention for their potential role in cancer — that are mutated with surprising frequency in the 3,281 samples in the dataset.
The researchers hope the new findings, published in Nature Genetics, will provide scientists with new leads in the search for somatic mutations that drive cancer. Additional data from the project, along with a downloadable version of the Hotnet2 software, is also available online.
“Ultimately, there will need to be laboratory experiments that confirm these findings,” said Ben Raphael, associate professor of computer science, director of the Center for Computational Molecular Biology at Brown, and the paper’s senior author. “But the hope is that the computational analysis will help prioritize the experiments toward those genes and mutations that are likely to be involved in cancer.”
The research takes a different approach than many cancer genetics studies, which often look for mutations in single genes that occur frequently in cancer samples. Genes often do not work alone, but operate together to form networks and pathways that govern cell functions. In some cases, a mutation in any of the multiple genes in a pathway could cause a malfunction that leads to cancer. Because damaging mutations can be spread across multiple such networks of genes, it can be hard to detect them in statistical tests.
“When looking at single genes, you typically find a small number that you can confidently say are likely to be cancer genes,” Raphael said. “But you also see many other genes that, statistically, you cannot say much about. You don’t know if they’re important or not.”
The Hotnet2 algorithm analyzes genes at the network level, and that helps to identify mutations that occur rarely but are nonetheless important in cancer.
“For example, maybe there’s a gene that’s mutated in 80 percent of samples, but the other 20 percent have rare mutations in multiple other genes,” Raphael said. “If we see that some of those rare mutations are in the same pathway as the more common one, it helps to build the case that those rare mutations are important.”
The HotNet2 algorithm works by projecting mutation data from patients onto a map of known gene interactions and looking for connected networks that are mutated more often than would be expected by chance. The program represents frequently mutated genes as heat sources. By looking at the way heat is distributed and clustered across the map, the program finds the “hot” networks involved in cancer.
The original version of Hotnet was used to identify networks important in acute myeloid leukemia, ovarian cancer, and several other types of cancer. Hotnet2 was modified from the original in order to deal with the much larger and more complex pan-cancer dataset used in this most recent study.
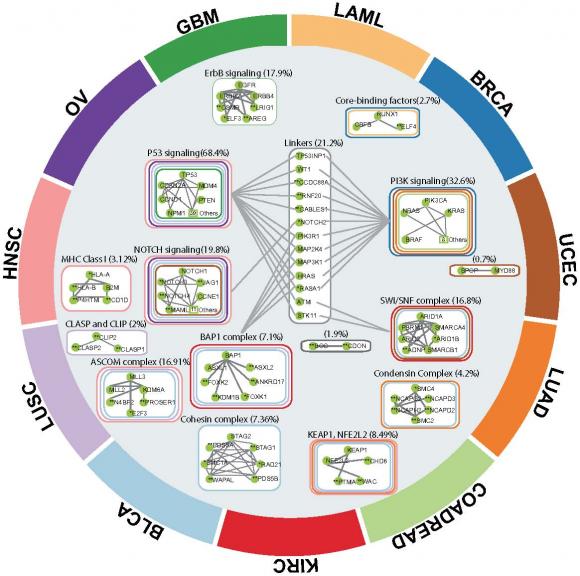
All told, the algorithm picked out 16 different networks that appear to be important across cancer types. Several of those 16 were networks associated with genes and pathways that are known cancer drivers, which provides a validation of the algorithm, Raphael said. Examples in that group include the p53 and NOTCH pathways.
But the algorithm also identified pathways that are not as well known as being important in cancer. Those included protein complexes such as cohesin and condensin, both of which play roles in cell division and other cellular processes.
Raphael hopes that research like this could point the way toward new laboratory investigations of these genes to confirm and better understand the role they may play in cancer. Ultimately, Raphael and his colleagues hope their network analysis will eventually help patients more directly.
“The next step is translating all of this information from cancer sequencing into clinically actionable decisions,” he said. “For example, there are now drugs that are used to treat patients who have mutations in particular genes. However, perhaps patients who don’t have a mutation in the targeted gene, but have a mutation in the same pathway, might respond to the same drug. This is the kind of analysis we would like to perform next.”
Max Leiserson, a student in Brown’s computational biology Ph.D. program and lead author of the study, is excited about the future of computational approaches to genetics and biology. “This type of analysis wouldn’t have been possible without the recent technological advances in both computing and DNA sequencing,” he said. “It is a very exciting time to be working in computational biology.”
In addition to Raphael and Leiserson, other Brown authors include co-first author Fabio Vandin, postdoctoral fellow Jason Dobson, graduate students Hsin-Ta Wu, Jonathan Eldridge, and Alexandra Papoutsaki, and undergraduates Jacob Thomas and Younhun Kim. They were joined by scientists from Yale University, University Pompeu Fabra, the Broad Institute, and the Genome Institute at Washington University in St. Louis.
“It was really a great team effort,” Raphael said
The work was supported by the National Science Foundation (grants IIS-1016648 and CCF-1053753) and the National Institutes of Health (grants R01HG005690, R01HG007069, R01CA180776, and U01HG006517).