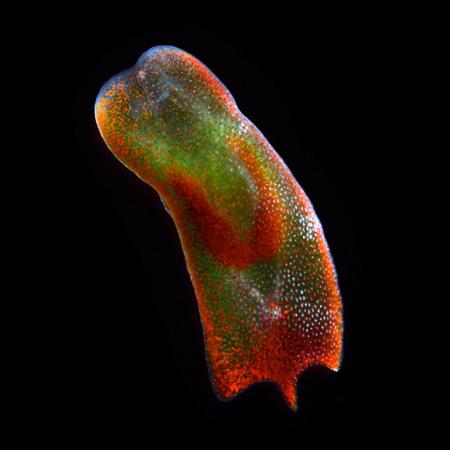
PROVIDENCE, R.I. [Brown University] — When it comes to understanding a critical junction in animal evolution, some short, simple flatworms have been a real thorn in scientists’ sides. Specialists have jousted over the proper taxonomic placement of a group of worms called Acoelomorpha. This collection of worms, which comprises roughly 350 species, is part of a much larger group called bilateral animals, organisms that have symmetrical body forms, including humans, insects and worms. The question about acoelomorpha, was: Where do they fit in?
To scientists, acoelomorpha, has been enigmatic, a “rogue animal,” said Casey Dunn, an evolutionary biologist at Brown University. “It has been wandering throughout the animal tree of life.”
The worm wanders no more. Through a laborious genetic sequencing analysis, Dunn and an international team of scientists have settled the long-standing debate and determined that acoelomorpha belongs as a sister clade to other bilateral animals. The finding is significant, Dunn said, because it shows the worm is a product of the deepest split within the bilateral animals, the first evolutionary divergence within the group. Because of that, scientists have gained a key insight into the most recent common ancestor to bilaterians, a species that remains unknown.
The worm is “as distant as an animal can be in bilateria and still be a bilaterian,” said Dunn, assistant professor of biology. “So, now we have two perspectives to (find out about) this common ancestor, the acoelomorphs and all the other bilateral animals.”
The results appear online this week in the Proceedings of the Royal Society B.
The team, composed of 17 scientists from the United States, France Germany, Sweden, Spain and the United Kingdom, had two more interesting findings:
- The debate appears to be over for Xenoturbella, a type of marine worm whose ancestral affiliation had been tossed between worms and mollusks. The researchers reported their genetic analysis shows diminishing evidence for placing xenoturbella within Deuterostomia, one of the major groups within the animal kingdom. Coincidentally, it also shows that xenoturbella may be a close relative to acoelomorpha.
- Cycliophora, a single species discovered in 1994 that lives on the bristles surrounding the mouth of the Norway lobster Nephrops norvegicus, has found a home with Entoprocta and Ectoprocta. The researchers base their findings on an analysis that reached further into the genetic makeup of cycliophora than previous studies had done.
The team used a genetic sequencing technique called expressed sequence tags to carry out the phylogenetic studies. The aim of this approach, discusssed in a study led by Dunn that appeared in Nature last year, is to analyze a large number of genes from a large number of animals. For this paper, the researchers looked at 1,487 genes, a 10-fold increase in the number of genes analyzed in previous studies. In all, the researchers logged 2.25 million processor hours on a supercomputer in California to obtain the results. Dunn called the effort the most computationally intensive phylogenetic analysis to date.
Other scientists who contributed to the research are Andreas Hejnol, Mark Martindale and Elaine Seaver, University of Hawaii; Matthias Obst, Goteborg University, Sweden; Alexandros Stamatakis and Michael Ott, Technical University of Munich, Germany; Greg Rouse, Scripps Institution of Oceanography; Gregory Edgecombe, Natural History Museum, London; Pedro Martinez and Jaume Baguna, Universitat de Barcelona, Spain; Xavier Bailly, Station Biologique de Roscoff, France; Ulf Jondelius, Swedish Museum of Natural History; Matthias Wiens and Werner Muller, Johannes-Gutenberg-University Mainz, Germany; Ward Wheeler, American Museum of Natural History; and Gonzalo Giribet, Harvard University.
The research was funded by the National Science Foundation and the San Diego Supercomputing Center.