The human genome is a dense jungle of genes. Five genes can be combined in more than 1019 ways. Even worse, scientists cannot look at the human genome in one piece and instead must look at fragments and then try to piece the genome together as if it were an immense jigsaw puzzle.
Ben Raphael, Eli Upfal, and Fabio Vandin in the Department of Computer Science had been working on various ways to allow biologists to locate mutating genes and to determine which of those mutations could lead to cancer — “to find those mistakes,” as Raphael put it. With ovarian cancer, there were dozens, possibly hundreds, of genes that could cause cancer. The trick was to find which genes partner with other genes to carry out the cancer-causing sequence.
“Instead of identifying individual players, can we identify the conspiracy?” said Raphael, associate professor of computer science at the Center for Computational Molecular Biology.
“That’s an algorithmic problem now,” Raphael said. “You have these individual genes, now identify the conspirators.”

But finding the target in any good conspiracy is never easy. So the team at Brown created a heat-seeker of sorts, which they call HotNet.
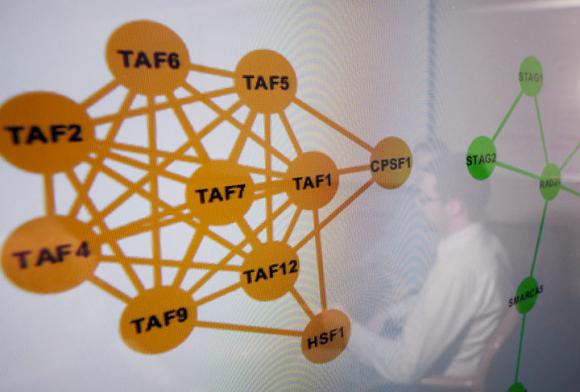
The algorithm treats mutated genes as heat sources and measures the intensity of the heat. When mutated genes cluster together, the “heat” generated by the group is more intense than the heat from a pair of mutated genes. This helps researchers to distinguish random mutated genes from concentrated groupings and to determine whether clusters “are other than random mutations,” Raphael said. “Are these hotspots in my data different than what I’d find with random data? Am I surprised by it?”
Vandin began developing HotNet as a class project as a visiting graduate student from the University of Padua, Italy. After earning his doctorate, Vandin returned to Brown as a postdoctoral researcher to continue refining the algorithm, supported by a grant from the National Science Foundation.
“I found the problem of identifying the significant mutations not only extremely important, but also challenging and fascinating,” Vandin said. “Looking at previous approaches, what was missing was a clever way to combine the interaction information with the mutation data and an efficient statistical test — two of the main features of HotNet.”
The paper, published in Nature in late June by the Cancer Genome Atlas Research Network, confirmed that mutations in a single gene — TP53 — are present in more than 96 percent of serous adenocarcinoma, the most prevalent form of ovarian cancer.
“Since TP53 is mutated in so many tumors, the gene is easy to identify on its own,” Raphael said. “HotNet is geared toward finding more subtle signals in the data that result from mutations across multiple genes. In the ovarian cancer study, HotNet pinpointed the Notch signaling pathway and the cohesin protein complex (involved in DNA repair) as significantly mutated. Neither of these was apparent by examining individual genes.”
Raphael said HotNet can be used to hunt for the genes responsible for causing other types of cancer. “We can use (the algorithm) in theory on any type of data, including non-biological,” he added.